Болезнь Вильсона-Коновалова – Патогенез

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
При болезни Вильсона-Коновалова имеется генетический дефект синтеза в печени церулоплазмина (медь-оксидазы), относящегося к а2-глобулинам. Значение церулоплазмина заключается в том, что он удерживает медь в крови в связанном состоянии. С пищей организм получает за сутки около 2-3 мг меди, в кишечнике примерно половина этого количества всасывается, поступает в кровь, связывается с церулоплазмином, доставляется к тканям и включается в специфические апоферменты.
Медь участвует в гемопоэзе, образовании костей. Небольшое количество меди находится в крови в ионизированной форме и выделяется с мочой.
При нарушении синтеза церулоплазмина увеличивается содержание в крови меди, не связанной с церулоплазмином, и она начинает откладываться в органах и тканях – печени, почках, головном мозге, поджелудочной железе и др. Этому способствует увеличение всасывания меди в кишечнике, что также наблюдается при этом заболевании. Накопление меди подавляет активность сульфгидрильных групп окислительных ферментов, нарушает тканевое дыхание, гликолиз и оказывает токсическое влияние на головной мозг.
Заболевание наследуется по аутосомно-рецессивному типу. Его распространённость составляет примерно 1:30 000, а частота носительства дефектного гена – 1:90. Ген болезни Вильсона расположен на длинном плече хромосомы 13, он клонирован и изучен. Ген кодирует переносящую медь АТФ-азу, с которой связывается 6 атомов меди. Местоположение в клетке и точная функция этого переносчика неясны. Возможно, он участвует в экскреции меди с жёлчью или в переносе её на церулоплазмин. В настоящее время при болезни Вильсона выявлено более 25 различных мутаций гена. Большинство из них приводят к изменениям скорее в функциональном домене АТФ-азы, чем в участках, связывающих медь. У многих больных мутацию идентифицировать не удаётся. Существует предположение, что при мутациях, приводящих к нарушению функционального домена, заболевание проявляется в более раннем возрасте. У большинства больных мутации на каждой из хромосом различны, что затрудняет установление соответствия между фенотипом и генотипом. Многообразие мутаций делает их исследование у отдельных больных с целью установления диагноза нецелесообразным.
Анализ гаплотипа, представляющий собой исследование аллелей маркёров-микросателлитов, расположенных вблизи дефектного гена на хромосоме 13, сыграл важную роль в установлении локуса этого гена. Однако и после клонирования дефектного гена этот анализ не утратил своего значения и применяется для исключения болезни Вильсона у братьев и сестёр больного или установления их гомо- или гетерозиготности по дефектному гену или нормы.
Это важно, поскольку у гетерозиготных носителей заболевание не развивается. Существует связь между гаплотипом и некоторыми мутациями, что может помочь в выявлении новых мутаций.
Крысы линии LEC (Long-Evans Cinnamon) являются естественной моделью для изучения болезни Вильсона. У них в течение первых нескольких месяцев жизни отмечаются значительное накопление меди в печени, низкий уровень церулоплазмина в сыворотке и развитие острого, а позднее и хронического гепатита. Эти изменения можно предотвратить назначением пеницилламина. В основе генетического дефекта у этих инбредных крыс лежит делеция гена переносящей медь АТФ-азы, который гомологичен гену болезни Вильсона.
Снижение экскреции меди с жёлчью при болезни Вильсона, а также в эксперименте на животных приводит к накоплению токсических количеств меди в печени и в других тканях. В результате перекисного окисления липидов происходит повреждение митохондрий, которое в эксперименте удаётся уменьшить с помощью витамина Е.
В норме у новорождённых значительно повышено содержание меди в печени и снижен уровень церулоплазмина в сыворотке. У новорождённых морских свинок содержание меди в тканях и уровень связывающего медь белка в плазме вскоре становятся такими же, как у взрослых особей. Остаётся неясным, связан ли этот процесс с изменением активности гена болезни Вильсона.
Степень изменений ткани печени может быть разной – от перипортального фиброза до субмассивного некроза и выраженного крупноузлового цирроза.
При гистологическом исследовании выявляют баллонную дистрофию и многоядерные клетки печени, скопления гликогена и гликогеновую вакуолизацию ядер гепатоцитов. Характерна жировая инфильтрация гепатоцитов. Клетки Купффера обычно увеличены в размерах. У некоторых больных эти изменения особенно ярко выражены; выявляются тельца Мэллори, что напоминает морфологическую картину острого алкогольного гепатита. У части больных наблюдаются изменения в печени, свойственные хроническому гепатиту. Гистологические изменения в печени при болезни Вильсона не являются диагностическими, однако выявление описанных выше изменений у молодых больных с циррозом печени позволяет заподозрить это заболевание.
Метод выявления меди окрашиванием рубеановой кислотой или родамином ненадёжен, поскольку медь распределяется неравномерно и в узлах регенерации отсутствует. Накопление меди обычно происходит в перипортальных гепатоцитах и сопровождается появлением атипичных отложений липофусцина.
Электронная микроскопия
Даже при бессимптомном течении заболевания выявляют аутофагические вакуоли и крупные изменённые митохондрии. Жировая инфильтрация может быть связана с повреждением митохондрий. Можно видеть инфильтрацию межклеточного пространства волокнами коллагена, а также светлые и тёмные клетки печени.
Поражение других органов
В почках выявляют жировые и гидропические изменения, отложение меди в проксимальных извитых канальцах.
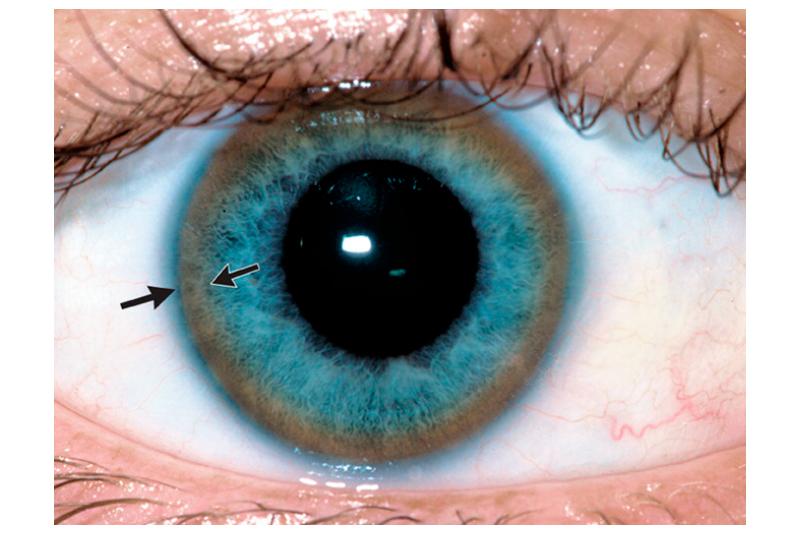
Кольцо Кайзера-Флейшера образуется при отложении содержащего медь пигмента в десцеметовой оболочке по периферии задней поверхности роговицы.
У некоторых больных эти изменения особенно ярко выражены; выявляются тельца Мэллори, что напоминает морфологическую картину острого алкогольного гепатита.
МКБ-10
- Причины
- Классификация
- Симптомы
- Диагностика
- Лечение болезни Вильсона
- Прогноз и профилактика
- Цены на лечение
Общие сведения.
Симптоматика
При болезни Вильсона-Коновалова симптомы в основном связаны с поражением печени (цирроз, печеночная недостаточность), нарушениями психики, ЦНС, комбинированными проявлениями. О том, что началась болезнь Вильсона, симптомы говорят не сразу. Лишь после достижения пятилетнего возраста у ребенка могут появиться первые ее признаки.

Симптомами являются желтуха, ощущение боли в области правого бока. Со временем происходит нарушение пищеварения.
Неоспоримым симптомом болезни Вильсона является желто-коричневое кольцо, расположенное на роговице (кольцо Кайзера-Флейшера). К этому присоединяются нарушения функции печени, почек, неврология и т.д.

При болезни Вильсона-Коновалова симптомы в основном связаны с поражением печени цирроз, печеночная недостаточность , нарушениями психики, ЦНС, комбинированными проявлениями.
Виды заболевания и их симптомы
В зависимости от клинических проявлений в медицинской практике выделяют три формы патологии Вильсона-Коновалова – болезнь, которая нарушает работу печени, заболевание, поражающее ЦНС, смешанный вид. Согласно типам у больного преобладает та либо иная клиника.
Гепатолентикулярная дегенерация сопровождается клиническим полиморфизмом, в процесс вовлекаются органы выделительной системы.
Патогенез [ править | править код ]
Медь выполняет множество функций в организме. В основном она выступает в качестве кофактора для некоторых ферментов, таких как церулоплазмин, цитохром с-оксидаза, дофамин бета гидроксилаза, супероксиддисмутаза и тирозиназа [4] .
Медь всасывается из желудочно-кишечного тракта. Транспортный белок на клетках тонкой кишки CMT1 (англ. Copper Membrane Transporter 1) перемещает медь внутрь клеток. Часть меди связывается с металлотионеином, а другая — перемещается в сеть Гольджи с помощью транспортного белка ATOX1. В аппарате Гольджи в ответ на повышение концентрации меди фермент ATP7A (англ. Copper-transporting ATPase 1) высвобождает этот элемент через воротную вену в печень. В печёночных клетках белок ATP7B связывает медь с церулоплазмином и высвобождает его в кровь, а также удаляет избыток меди с выделяющейся жёлчью. Обе функции ATP7B нарушены при болезни Вильсона. Медь накапливается в ткани печени; церулоплазмин продолжает выделяться, но с недостатком меди (апоцерулоплазмин) и быстро разрушается в кровотоке [4] .
Когда меди в печени становится больше, чем белков её связывающих, происходит их окислительное повреждение за счёт реакции Фентона. Это приводит к воспалению печени, её фиброзу и в итоге к циррозу. Также из печени в кровоток выделяется медь, которая не связана с церулоплазмином. Эта свободная медь оседает по всему организму, особенно в почках, глазах и головном мозге.
Основную роль в патогенезе играет нарушение обмена меди, её накопление в нервной (особенно поражены базальные ганглии), почечной, печёночной ткани и роговице, а также токсическое повреждение медью данных органов. Нарушение метаболизма выражается в нарушении синтеза и снижении в крови концентрации церулоплазмина. Церулоплазмин участвует в процессе выведения меди из организма. В печени формируется крупноузловой или смешанный цирроз. В почках в первую очередь страдают проксимальные канальцы. В головном мозге поражаются в большей степени базальные ганглии, зубчатое ядро мозжечка и черная субстанция. Отложение меди в десцеметовой мембране глаза приводит к формированию кольца Кайзера-Флейшера.
Во многих случаях появлению симптомов поражения нервной системы предшествуют висцеральные расстройства в виде нарушения деятельности печени и желудочно-кишечных расстройств желтуха, боли в правом подреберье, диспептические явления.
402 Группы – Круглов Степан Сергеевич
Преподаватель: Зуев Андрей Александрович
Болезнь Вильсона-Коновалова
Болезнь Вильсона-Коновалова (или гепатоцеребральная дистрофия) – редкое наследственное заболевание, в основе которого лежит генетически обусловленное нарушение обмена меди с избыточным ее накоплением преимущественно в печени и нервной системе. Описано в 1883 г. Вестфалем и в 1912 г. Вильсоном. Термин «гепатоцеребральная дистрофия» предложен Н.В.Коноваловым.
Этиология и патогенез.
В основе лежит аутосомно-рецессивное наследственное нарушение метаболизма меди; ген расположен в длинной части хромосомы 13. Распространенность в различных регионах мира в среднем 1:30000 при частоте гетерозиготного носительства около 1 %.
Первоначально ген экспрессируется в печени, почках, плаценте. Продукт гена представляет собой катионтранспортирующий Р-тип АТФазного протеина. Следствием генетического дефекта является различной степени выраженности нарушение функции внутриклеточного транспорта меди. Это ведет к снижению экскреции меди с желчью и накоплению её в гепатоцитах.
С пищей в сутки поступает 2-5 мг меди. Она всасывается в кишечнике, поступает в печень, где связывается с синтезируемым печенью церулоплазмином, циркулирует в сыворотке крови, избирательно захватывается органами и экскретируется с желчью.
В норме экскреция меди с желчью 2 мг в сутки, при болезни Вильсона-Коновалова – только 0.2-0.4 мг, что приводит к повышенному накоплению меди в организме.
Включение меди в церулоплазмин происходит в аппарате Гольджи при участии гена гепатоцеребральной дистрофии. Незначительная часть меди находится в крови в ионизированной форме в виде лабильного комплекса с альбумином и выделяется с мочой.
При болезни Вильсона-Коновалова увеличена абсорбция меди в кишечнике, снижена экскреция меди с желчью. Снижение экскреции меди связано с дефектом гена гепатоцеребральной дистрофии, определяющего транспорт меди в аппарат Гольджи и последующее выделение лизосомами в желчь. Нарушается процесс включения меди в церулоплазмин. Из-за недостаточного использования меди происходит её депонирование в печени, мозге, почках, роговице. Депонированная в печени медь вторично ингибирует синтез церулоплазмина.
Уровень церулоплазмина в сыворотке крови имеет диагностическое, но не патогенетическое значение. У 5 % больных определяется нормальный уровень церулоплазмина. При биопсии печени у таких больных имеется избыточное количество меди, также увеличивается содержание меди в крови и тканях, выделение её с мочой.
Медь, являясь прооксидантом, оказывает токсическое действие на организм. Её накопление ведет к повышенной продукции свободных гидроксильных радикалов. При обследовании больных болезнью Вильсона-Коновалова и животных с экспериментальной перегрузкой медью в плазме крови определяется снижение уровня витамина Е, увеличение циркулирующих продуктов перекисного окисления липидов; в печени снижены уровни восстановленного глутатиона и витамина Е.
Митохондрии печени являются мишенями действия оксидантов. Нарушение дыхательной цепи и снижение активности цитохром-С-оксидазы увеличивает продукцию свободных радикалов благодаря утечке электронов из дыхательной цепи.
Свободная медь, накапливаясь в тканях, блокирует SH-группы ферментов, участвующих в окислительно-восстановительных реакциях. Это приводит к энергетическому голоданию, к которому наиболее чувствительна ЦНС.
В начале заболевания, когда клинические признаки отсутствуют (I стадия), медь накапливается в цитозоле печеночных клеток. Медь, связанная с SH-группами цитозольных протеинов, затрудняет секрецию гепатоцитами белков и триглицеридов. Наступает стеатоз гепатоцитов и появление телец Маллори.
Во II стадии медь перераспределяется из цитозоля в лизосомы гепатоцитов. Часть поступает в кровь. В связи с низкой специфической активностью лизосом билиарная экскреция меди понижается. Медь вызывает переокисление липидов и повреждение лизосомальных мембран с последующим выходом вредных кислых гидролаз в цитоплазму. Наблюдаются некроз гепатоцитов, развитие хронического гепатита и гемолитической анемии.
В III стадии усиленное накопление меди в печени приводит к фиброзу и циррозу. Повышенное накопление меди в головном мозге, роговице, дистальных отделах почечных канальцев приводит к развернутой картине болезни.
В печеночной ткани наблюдаются жировая дистрофия гепатоцитов, перипортальный фиброз, субмассивные некрозы гепатоцитов, макронодуллярный цирроз. В почках – жировая и гидропическая дистрофия с отложением меди в проксимальных канальцах.
Нарушение дыхательной цепи и снижение активности цитохром-С-оксидазы увеличивает продукцию свободных радикалов благодаря утечке электронов из дыхательной цепи.
Болезнь Вильсона
Болезнь Вильсона — это заболевание, передающееся по наследству, его причина заключается в нарушении накопления и транспорта меди в организме, вследствие чего происходит хроническая интоксикация. В крови понижается концентрация белка, который транспортирует медь, церрулоплазмина. Полное название болезни звучит как Болезнь Вильсона-Коновалова, гепатоцеребральная дистрофия, гепатолентикулярная дегенерация, болезнь Вестфаля-Вильсона-Коновалова.
- Классификация болезни Вильсона
- Этиология и патогенез болезни Вильсона
- Клиническая картина болезни Вильсона
- Диагностика болезни Вильсона
- Лечение болезни Вильсона
- Прогноз при болезни Вильсона
- Профилактика болезни Вильсона
Патология провоцирует множественные наследственные заболевания внутренних органов и центральной нервной системы, особенно печени и промежуточного мозга. Передается это заболевание по аутосомно-рецессивному типу. Патологический ген ATP7B (находится в 13 хромосоме) мутирует, что и вызывает нарушения.
В случае, если человек получает дефектный ген от обоих родителей, болезнь начинает проявляться и прогрессировать. Человек, имеющий один патологический мутантный ген является носителем заболевания, концентрация меди отклонена от нормы, но нарушения незначительны.
Диагностирование заболевания приходится на возраст 10-13 лет. Болезнь проявляет себя как смешанный цирроз печени или почечная недостаточность. Чаще болеют мальчики. Распространённость болезни Вильноса повышается среди народностей, у которых популярны браки между кровными родственниками.
Клиническая картина болезни Вильсона.
Лечение
Целью лечения болезни Вильсона-Коновалова является уменьшение поступления меди с пищей, а также уменьшение запасов меди в организме. Для этого назначаются соответствующие лекарственные препараты и специальная диета.
Некоторое положительное значение имеет назначение антигистаминных препаратов, витаминов, а также препаратов, уменьшающих сосудистую атонию и проницаемость.
Лечение гепатолентикулярной дегенерации является пожизненным.
Выраженные случаи болезни Вильсона-Коновалова не представляют больших диагностических трудностей.
Характеристика и формы
Болезнь Коновалова-Вильсона известна так же как гепатоцеребральная дистрофия. В клинической практике болезнь также называется гепатолентикулярная дегенерация, которая связана с метаболическими нарушениями, в частности со сбоями в процессе обмена меди. Распространенность патологии составляет около 5 случаев на 300-400 тысяч населения. Статистика показывает, патология чаще выявляется среди мужчин.
Для болезни Вильсона-Коновалова типично несколько вариантов развития, что отражается на клинике, в связи с чем различают формы – печеночную и неврологическую. Доля пациентов с печеночной формой составляет 50-80%. При печеночной форме происходит прогрессирующее поражение печени, что приводит к развитию гепатита, в тяжелых случаях – цирроза печени. При неврологической форме в клинической картине преобладают симптомы, связанные с нарушением работы ЦНС.
В 20% случаев заболевание дебютирует психическими расстройствами. Неврологическая симптоматика появляется как дебютный признак в 40% случаев. Некоторые клиницисты выделяют смешанную (проявляется неврологическими и печеночными симптомами) и бессимптомную форму, которая до определенной стадии не проявляется выраженными нарушениями. В перечне МКБ болезнь Вильсона-Коновалова значится под кодом «E83».
Для печеночной формы характерно бессимптомное длительное течение.
Что это за болезнь
13 хромосома содержит тот ген, который вызывает развитие патологии. С его помощью медь транспортируется в желчь и включается в церулоплазмин (белок, содержащийся в крови). Таким образом, причины болезни Вильсона-Коновалова являются генетическими.
Заболевание передаётся по наследству по аутосомно-рецессивному типу наследования. Оно возникает, если оба родителя передают дефектный ген.
Нормой содержания меди в организме здорового человека считается не более 100–150 мг при суточной потребности в 1–2 мг. Если же наблюдается избыток, то он выводится из печени в кровь или с помощью желчи. Количество белка, связывающего медь в крови (церулоплазмина), при этом уменьшается.
В результате медь поступает в свободном виде в кровь и к другим органам, начинает откладываться в них. Наибольшая концентрация наблюдается в печени, почках, нервной системе. Отмечается токсическое поражение этих органов. На роговице глаза возникает так называемое кольцо Кайзера-Флейшера: коричневатое отложение меди по ободку зрачка.
Существует единственная причина, а именно мутация гена.
Лечим печень
У больных с экстрапирамидной патологией могут развиваться пирамидные моно- и гемипарезы.
Болезнь Вильсона-Коновалова

И.А. Иванова-Смоленская
профеcсор, доктор медицинских наук
ГУ НИИ неврологии РАМН
Болезнь Вильсона–Коновалова (гепато-лентикулярная дегенерация) относится к тяжелейшим наследственным болезням центральной нервной системы и внутренних органов. Если своевременно не начать лечение, направленное на выведение токсичных избытков меди из организма, то через 5–7 лет больной обречен на смерть. Болезнь поражает 25% братьев и сестер в семье при клинически здоровых родителях, которые являются носителями аномального гена (аутосомно-рецессивный тип наследования). Заболевают только те индивидуумы, которые унаследовали два мутантных гена, то есть по одному от матери и от отца – гомозиготные носители мутации; лица, которые от одного из родителей получили мутантный ген, а от другого – нормальный ген, являются гетерозиготными носителями мутации и остаются здоровыми.
Открытый недавно ген болезни отвечает за синтез медь-транспортирующего белка (АТР7В). При гепатолентикулярной дегенерации обмен меди и медьсодержащих белков нарушается, появляется избыток “свободной” меди, которая в больших количествах откладывается в печени, мозге, роговице, а также выделяется с мочой. Не случайно диагностика болезни базируется на обнаружении характерных нарушений медного обмена. Благодаря идентификации гена в настоящее время возможна и ДНК- диагностика этого заболевания.
Поражение печени избытком “свободной” меди проявляется циррозом печени. Поражение мозга приводит к развитию тяжелой неврологической симптоматики: дрожанию конечностей и всего туловища, повышению мышечного тонуса, иногда сопровождающемуся болезненными спазмами, нарушением речи, глотания, снижению интеллекта. Отложение меди в роговице (по краю радужной оболочки) обусловливает формирование кольца Кайзера–Флейшера – буро-зеленоватого пигмента. По этому признаку диагноз болезни можно поставить безошибочно.
Гепато-лентикулярная дегенерация известна с глубокой древности. Дошедшее до нас изображение египетского фараона Тутанхамона, по мнению крупнейшего специалиста J. Walshe, не исключает вероятности, что он страдал этим заболеванием. Институт неврологии РАМН в течение многих лет занимается проблемой гепато-лентикуляной дегенерации. Знаменитый отечественный невролог академик АМН СССР Н.В. Коновалов, один из основателей Института, посвятил этому заболеванию две монографии, последняя из которых в 1964 году была удостоена Ленинской премии. В последующие годы данное заболевание продолжало успешно изучаться сотрудниками нейрогенетического отделения Института неврологии, под наблюдением которых за 40 лет находилось свыше500 семей, отягощенных этим недугом. Весь многолетний опыт Института свидетельствует о том, что ключевой проблемой является ранняя диагностика гепатолентикулярной дегенерации. Чем раньше начать лечение (в идеале – еще на досимптоматической стадии либо на доневрологическом этапе, то есть до появления признаков поражения мозга), тем лучше эффект. Вот почему, если в семье есть хоть один ребенок, страдающий этим заболеванием, необходимо тщательное обследование всех его братьев и сестер, в том числе с использованием самых современных биохимических и молекулярно-генетических методов.
Заподозрить раннюю стадию болезни можно на основании следующих признаков: перенесенной желтухи; повторных кровотечений из носа, кровоточивости десен либо множественных кровоподтеков; сосудистых “звездочек” на коже груди и спины; своеобразных “полосок” (белых, меняющих периодически окраску на красновато-синюшную) на бедрах и в подмышечных областях; гормональных нарушений в виде аменореи или дисменореи у девушек, гинекомастии (нагрубание грудных сосков) у юношей, а также акромегалии(увеличение носа, подбородка, утолщение губ); снижения интеллекта и изменений психики в виде чередования дурашливости и пониженного настроения, трудностей усвоения нового материала, проблем с успеваемостью в школе.
Гепато-лентикулярная дегенерация может начать проявляться в детском, подростковом, юношеском, зрелом возрасте и очень редко – в 50–60 лет. Чем раньше начинается заболевание, тем тяжелее оно протекает (при отсутствии лечения). Однако болезнь Вильсона–Коновалова – редкий пример наследственного нарушения, для которого разработаны высокоэффективные методы лечения: даже при появлении тяжелой неврологической симптоматики систематическое лечение обычно дает “драматический” эффект, вплоть до исчезновения всех симптомов или резкого их уменьшения. Пациенты вновь могут полностью обслуживать себя, вести домашнюю работу, учиться, работать по профессии, создать семью и родить здорового ребенка (под нашим наблюдением находятся 30 женщин, страдающих гепатолентикулярной дегенерацией и благополучно родивших здоровых детей). Пациентам с гепатолентикулярной дегенерацией необходимо регулярно наблюдаться у постоянного лечащего врача.
В чем же заключается лечение этой тяжелейшей болезни? Во-первых, это строгое соблюдение “печеночной” диеты (стол 5а), предполагающей исключение богатых медью продуктов (шоколад, кофе, орехи, бобовые и др.). Однако основное лечение – постоянный прием препаратов, выводящих медь из организма. Главным из них является D-пеницилламин.
Эти препараты назначаются по специальной схеме с постепенным увеличением дозы. К сожалению, в силу необходимости проведения пожизненного лечения и особых требований к химической чистоте препаратов отечественный аналог пеницилламина не может быть рекомендован при гепатолентикулярной дегенерации из-за высокой токсичности.
При длительном многолетнем приеме D-пеницилламина у некоторых больных гепатолентикулярной дегенерацией возникают побочные явления в виде дерматитов, анемии и иных осложнений. Поэтому был предложен альтернативный метод лечения солями цинка (оксид, сульфат и др.). Нами было предложено комбинированное лечение D-пеницилламином и препаратами цинка, что дает возможность снизить дозу и избежать побочных явлений. У больных в пресимптоматической стадии достаточно лечения только препаратами цинка.
В настоящее время за рубежом в тяжелых случаях болезни, не поддающихся консервативному лечению, широко применяется пересадка печени. При удачном исходе операции больной полностью выздоравливает и не нуждается в дальнейшем приеме препаратов. В России делаются первые шаги в этом направлении, и одним из таких шагов является разработанный нами совместно с Институтом трансплантологии и искусственных органов метод био-гемоперфузии с изолированными живыми клетками печени и селезенки – так называемый аппарат “вспомогательная печень”.
Помимо этих методов, большое значение имеет гепатопротекторная терапия, направленная на максимальное улучшение функций печени.
Таким образом, при правильной терапии гепатолентикулярной дегенерации – тяжелейшего наследственного заболевания мозга и внутренних органов – в 80% случаев возможно клиническое выздоровление либо выраженное улучшение состояния больных при условии своевременной максимально ранней диагностики.
В последующие годы данное заболевание продолжало успешно изучаться сотрудниками нейрогенетического отделения Института неврологии, под наблюдением которых за 40 лет находилось свыше500 семей, отягощенных этим недугом.
Роль меди в норме и при гепатолентикулярной дегенерации
Здоровый человек получает ежедневно с пищей 2 мг меди, из них усваивается только 1/3 часть, но и этого вполне хватает для обеспечения потребности организма.
Медь необходима печени:
- для синтеза белков и ферментов, а значит для роста, физического и умственного развития;
- образования гемоглобина, транспорта железа;
- синтеза эритроцитов и лейкоцитов;
- обеспечения эластичности стенок сосудов за счет участия в синтезе коллагена;
- стимуляции гормональной активности гипофиза и инсулина;
- выработки клеток иммунитета;
- образования антиоксидантов, предотвращения раннего старения.
Попадая их тонкого кишечника в кровь, по воротной вене молекулы меди доставляются в печеночные клетки. Здесь часть связывается с белком церулоплазмином и возвращается в кровяное русло, другая (лишняя) — переходит в желчь и удаляется из организма. Одна молекула церуллоплазмина включает 8 атомов меди.
В патогенезе болезни Вильсона-Коновалова нарушаются обе функции. Церулоплазмин разрушается. Кровь больного переполняется значительным количеством несвязанной меди. Она откладывается в разных органах: в печени, головном мозге, почках, роговице глаз, переходит в мочу.
В гепатоцитах печени, как ответная реакция на внедрение меди образуется воспаление, переходящее в узловой цирроз. В почечной ткани страдают проксимальные канальцы. Повреждение головного мозга выражается в поражении базальных ганглиев, ядер мозжечка и черной субстанции.
В тканях глаза медь откладывается вокруг роговой оболочки и образует видимое кольцо Кайзера-Флейшера. Преимущественное отложение меди в одном из органов зависит от возраста. Для детей типично поражение печени. После 20 лет основные разрушения локализуются в головном мозге.
Среди неврологических признаков.
Глава 2.: «Болезнь Вильсона-Коновалова»
 Книга: «Редкие неврологические синдромы и болезни» (В.В. Пономарев)
Книга: «Редкие неврологические синдромы и болезни» (В.В. Пономарев)
Назначен купренил до 6 табл сут.
Лечение болезни Вильсона-Коновалова
Лечат заболевание при помощи:
- лекарств, удаляющих медь из организма, называемых хелатирующими агентами;
- цинка, препятствующего поглощению меди кишечником.
Во многих случаях лечение улучшает или предотвращает симптомы и повреждения органов. Врачи также могут порекомендовать изменить диету, чтобы избежать употребления продуктов с высоким содержанием меди.
Люди с болезнью Вильсона-Коновалова нуждаются в пожизненном лечении. Прекращение терапии может привести к острой печеночной недостаточности. Медицинские работники регулярно будут проводить анализы крови и мочи, чтобы проверять, как протекает лечение.
Лечение болезни Вильсона-Коновалова.





